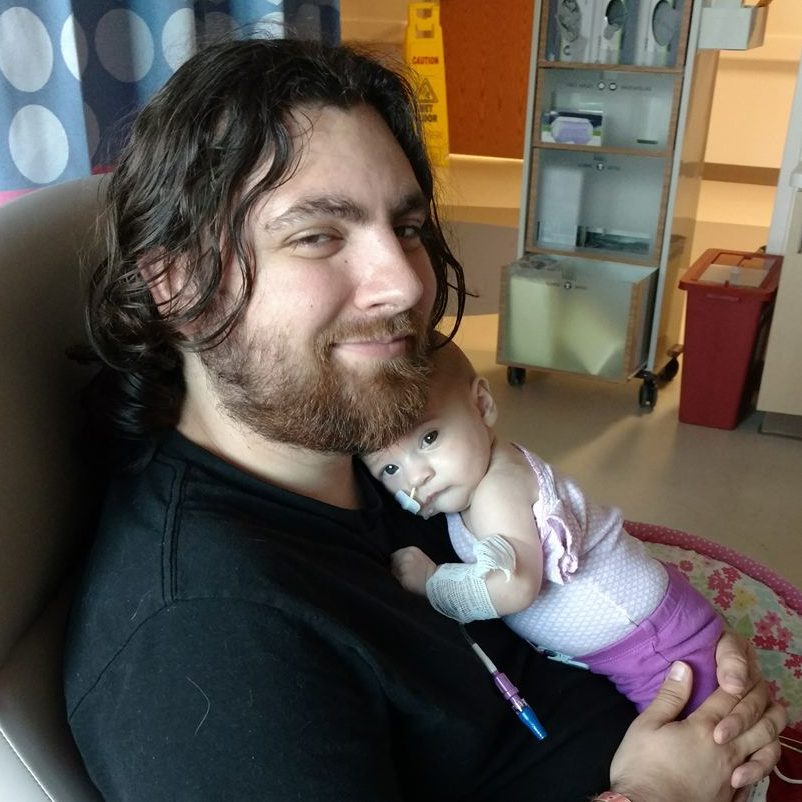
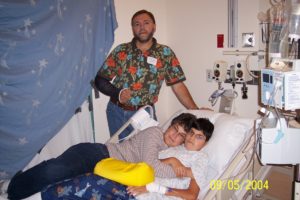
It’s a scene I’m very familiar with. The parents sit across from me, anxious and fidgety. Sometimes they cry, sometimes nervous giggles. Always fear. Confusion and concern for their child. As a psychiatric nurse, I know the parents are, by extension, secondary patients that I must also care for. I help calm them down as they try to rationalize their child’s attempt, or most recent self-harm. I reassure them that this isn’t the end of the world. There is hope for them and for their child. I help them come to grips with their loss of control. I explain the treatment, sometimes I can explain the behavior. Sometimes it is more normal than they realize. Patient care is something I’m fairly familiar with; it is easy to comfort from the outside looking in.
But then, it’s your own child…and everything is different.
You realize your hypocrisy. All those times you reassure and quiet their fears, only to spiral in panic as soon as you are placed in their shoes. There is a realization that hits in, how little control you have, and how truly helpless you are. You are simply a silent observer, through tear-stained glasses, forced to watch a tragic performance, deviating from a script you didn’t write.
Every lab draw was agony. Every IV and test broke me. Even the feeding tubes and consistent weight loss. Almost every procedure was something I had done before or observed. I never imagined during my training that I would see my own daughter submitted to the same. All of my experience and training was useless. Everything is different when the patient is your child.
I remember holding my precious girl’s little hands, listening to the doctor explain, again, how they didn’t know what to do. I prayed silently, hoping someone could hear me. So much uncertainty. Every test and every potential answer was a red herring. I didn’t know how much longer I would be able to refer to my daughter in the present tense. That question burned in my head every time I visited the NICU.
I finally interrupted the doctor to ask as bluntly as I dared. “Will my baby die?” The doctor replied through tears that she did not know. I left feeling nothing. I had no more tears to give and no strength to feel sorrow. I had to accept that my daughter would probably never come home.
That type of pain damages you. In some ways, you do not fully recover. It is almost as if part of your soul dies in that moment. I cannot begin to imagine the pain of actually losing your child. My daughter pulled through…but there is still a large scar in my heart. It will haunt me, forever.
It was so difficult to accept that my daughter would live. Almost more difficult than accepting that she wouldn’t. Answers came, and with them, solutions…but none of it felt real. It took a while for me to hope again. To let myself plan a future. To assume she would be OK.
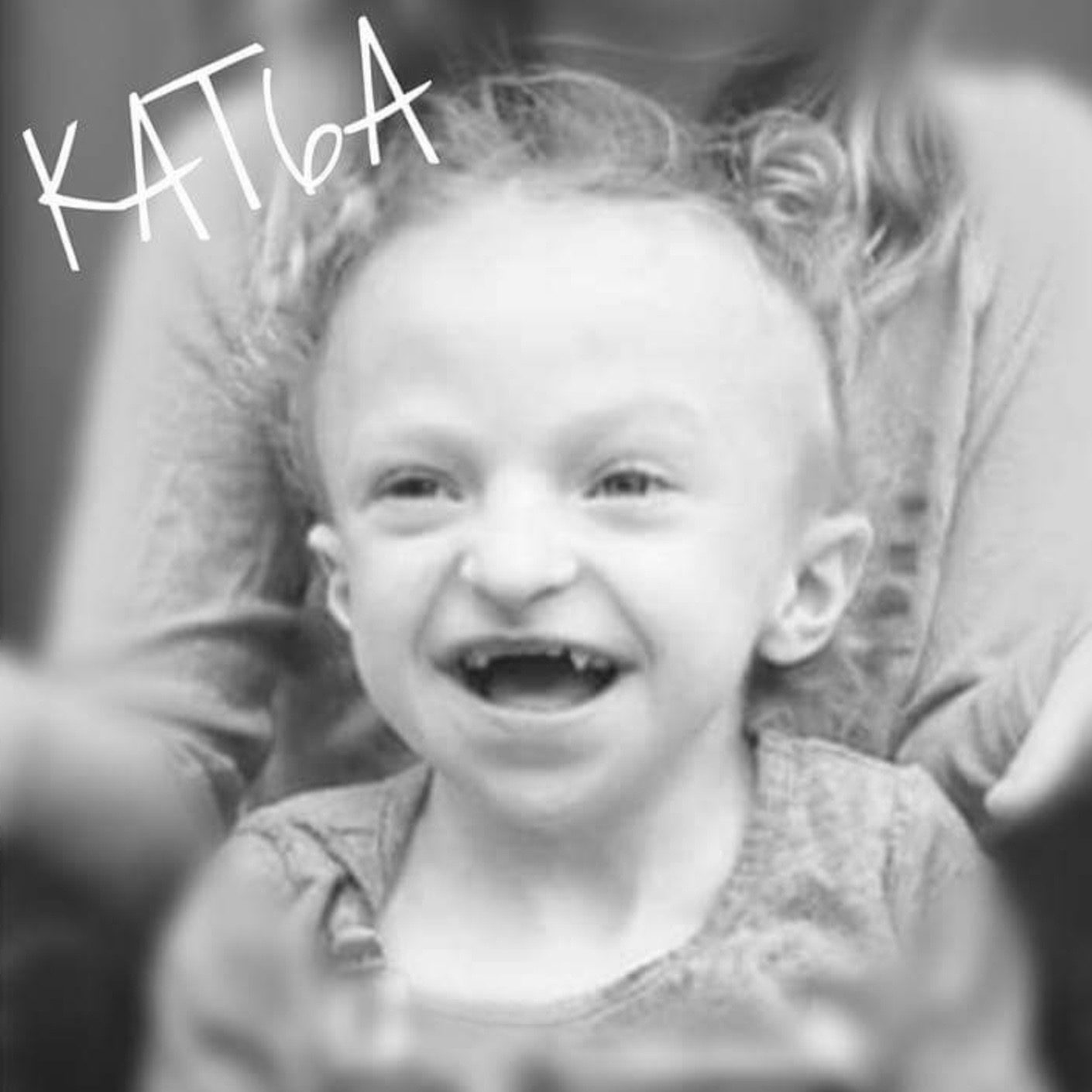
I did, though. My daughter, diagnosed with KAT6A, is developmentally delayed…but healthy, living, and progressing. We are a part of a support group, and going to a clinic specifically for KAT6A in a couple weeks. Her story isn’t over.
At this point, I would love to end with a cheesy message of hope. But I have none. Only a message of pain. But pain can connect people, and strengthen bonds. Pain bares open our souls and reveals our truest self.
If you are reading this, you have likely experienced pain. Perhaps you, like me, have had to watch your loved one suffer. Just know that I, too, feel your pain. I do not understand it. Yours is unique to you, and I will never claim to know how you feel, but know you do not suffer alone. We all carry our own burdens, often too heavy to hold on our own. I share my pain with you. If it somehow resonates with you, please consider sharing your own pain. Allow us to share your suffering. You do not need to hurt alone.
-Tobias
Filed under: Blog
Comments: Comments Off on Both Sides of the Bed
Peter is a nine-year-old boy diagnosed with KAT6A. Despite being nonverbal he is literate and enjoys typing on his keyboard. Below you can read his personal blog. His blog entries are unedited and will be updated frequently.
January 16, 2018
Hi kat6a parents,
My name is Peter. I have many disabilities. I like to dance, read books, hear happy songs and ride my bike. I feel happy because my family is always trying to find a cure for kat6a. I like to color rainbow. I like to watch true stories. The universe is full of bogus yet no one minds. The goal of my life is to joyfully teach human history.
Love,
Peter
January 18, 2018
My experience with skiing is truly unique. I established a real understanding of the height of the hill. I feel joyful when I go down the green hill. The lift is dangerous because the feet don’t touch the floors. I like the magic carpet because it’s the safest real way to enjoy skiing.
January 23, 2018
Greetings from the rare disease foundation,
I want to thank everyone for the happy threads. I am delighted to read your joyful replies. I would love to meet Sam, I want to try hiking in Maine and to eat fish.
January 29, 2018
Regards from West Nyack,
I am grateful for the joyful reward received at the talent show. I had enjoyed the great experience, joy, reaction of the people and the great responses.
I believe that i never saw a moose. I personally hate that the extinction of the moose will tremendously ruin the earth. I truly would like to see a moose.
February 9, 2018
Hi from your friend Peter,
Riding the lift was truly rewarding. I experienced the height of the blue hill, green hill, orange hill and understood the dimensions. I love to hike in the iced forest. I read books to explain our true happiness, real joys and ultimate rewards. Life is very erratic. The people I know try to find happiness.
I really enjoyed meeting all the kat6a families, doctors and the excellent subjects. I really enjoyed the interactions between the youngest kids, researchers and the experience of imagining myself being a renowned innovator.
February 12, 2018
Hello,
I enjoyed playing the piano at the talent show off West Nyack Elementary School . I liked the unexpected support of the teachers and friends. I hope that I’ll be able to talk next year because i would love to sing the national anthem. Hearing the children joyfully singing was rewarding. I’m hungry now so I’ll go to the kitchen to eat lunch.
Hugs from Peter
February 21, 2018
Dear friends,
The true treasure is happiness. I’m impressed by people understanding the meaning of the joyful universe. I want to tell my kat6a friends that reading is important because it helps understsanding real events.
Reading about the killing of the moose is very upsetting. I really understand the true reason of the hunting.
Regards
Peter
March 2, 2018
Dear friends,
The weather is fluctuating. The temperatures rise to a high level then drop to extreme cold.
The vision of the respectful award from NYS PTA truly made me happy. I m frustrated because reading aloud is very hard for me. I wish reading was easier. I try to express my thoughts by typing.
March 15, 2018
Dear readers,
I was trapped for over 2 hours in a ski lodge at mount peter in warwick, new york during the snowstorm on march 7. The tow truck towed the car, the driver really exceeded my expectations by taking us and the car to the motel. I ate Chinese food and slept with my family at the motel. I was relieved when I got home.
I hate being trapped because I feel fragile. I like hiking when it’s warmer.
Joy to all
March 23, 2018
Dear friends,
The weather exceeded my expectations. It’s snowing in the spring, the schools are closing and it has been boring. I like my teacher, she comes home and sits in my room to teach me. I heard that June will be warm, the pool will be open and there will be a town festival. I really love hiking, swimming and riding my bike. I remember reading a magazine about hiking in the catskills. I wish I can go there.
April 23, 2018
Dear enthusiastic readers,
I like to enjoy nature in winter and spring. The woods are pretty. I hear the birds expressing happily their feelings. The trees are turning green. Reading empowers young children. Reading makes a difference for children who can’t talk. Reading experiments greatly helps researchers doing their research.
May 3, 2018
Dear readers,
My brother Paul had his first communion the last weekend. We went to church then to brunch. We had the best brunch. Then, we went to the pool.
Sunday was busy too. We had great time at six flags. I went on most of the rides. It was very fun.
May 30, 2018
Dear readers,
I’m extremely happy to be playing the piano on Sunday. I never thought that I’ll be able to play this level. I wish I can talk more. I’m very hopeful that research reaches real outcomes.
June 12, 2018
Dear friends,
I’m very happy for my brother, John, who just finished preschool. He loves to read Scooby Doo books. John is very smart. He reads and plays good on the piano. The weather is nice. I’m riding my bike every day. The joyful rhyme of the birds truly reassures me. I love observing the nature in the spring.
I wish you all a happy summer.
July 15, 2018
Dear friends,
The weather has been nice, I’m going to the Germonds pool every afternoon. I love
the big pool because I can swim laps, greet people, try to play with friends, grant eventual friends
deserts and enjoy the clean water. Germonds pool is my favorite place during the summer. Real
humans come to this pool, they believe in my abilities and treat me with respect.
August 14, 2018
Dear readers,
I really try to behave but I can’t always control my behaviors. I like to go to the pool every day. Swimming helps me control my impulsivity. I deserve to go to school. I’m trying to do my best so my teacher takes me to school. I love to ride on the bus.
Peter
October 1, 2018
Dear kat6a friends,
Healing from bacterial infections takes very long time. My throat was hurting for fifteen days. I got cefdinir and i drastically felt better. I’m thankful for my doctor and my family. I joyfully read your nice comments.
October 23, 2018
Dear friends,
I turned 10 last Sunday. I loved my lattice pie. My family is amazing. Everyone made my day special. I miss going to school. I’m hardly trying to control my impulsivity but I’m not always successful.
November 14, 2018
Dear friends,
I like the fall. The leaves are crispy and have numerous colors. I was raking the leaves really hard when it started to rain. I had to escape the rain and hide under a roof. Hearing the rain was great.
January 4, 2018
Dear friends,
I restarted home instructions this week and it has been a totally boring end of week. Reading requires high attention and focus. I enjoy doing mathematics and science. I hope you all had a nice new year’s eve celebration.
On April 29, 2019, for the second year, Peter was honored with PTA’s Reflections Winner of Literature. Below is his essay entry.
Heroes Among Us
by Peter Najm
Real heroes make the world joyful and safe. Heroes are among us, between us and with us. We are all heroes. We are heroes by being nice. We are heroes by not bullying. We are heroes by not fighting. We are heroes by being humble.
Heroes are real. Brave firefighters are heroes. Smart doctors are heroes. Dedicated teachers are heroes. Humble professors are heroes. Fair judges are heroes. Honest leaders are heroes. Excellent chefs are heroes.
My sister, Mary, is a hero. She tries to play games that older kids play. She likes the high rollercoasters, giant rides and carousels.
My brother, John, is a hero. He reads frightening books and is not scared. He skis very fast fiercely.
My brother, Paul, is a hero. He manages to play with children of all ages. He behaves really good at church.
My mom, Natacha, is a hero. She works at night as a doctor. During the day, she teaches me and my siblings sports, French, good behaviors and helps us with general homeworks.
My dad, Emile, is a hero. He is an engineer, handyman and a musician. He doesn’t take a break. He always find ways to teach me. I’m very hyperactive and impulsive.
Real people try to become heroes. They want to impress their mothers, their fathers, their directors and their friends. They receive honors if they highly perform verbally, by writing, by dancing, by acting, or by volunteering.
Filed under: Blog
Comments: Comments Off on Peter’s Personal Blog
When parents have a baby or child with a serious diagnosis, there are many unknowns. One is the future—what will happen as my child grows up. Will his siblings resent all the attention I give him? Will he have friends? Will he have a place to belong? Will his life have meaning? Will I be able to meet his needs? Will that be enough? Are all our dreams of the future shattered?
Here is Sam’s story.
Sam was born in August 1990 six weeks prematurely. Nothing was right, even from the start. He didn’t breathe, he couldn’t regulate his body temperature, and he couldn’t eat. And things didn’t get too much better very fast. He had bradychardia, he had strange breath-holding spells (or were they seizures?) when he would turn blue and pass out. He spit up a lot, he had terrible screaming spells, he had pneumonias, he wasn’t moving normally, his developmental milestones weren’t being met…he was blind. That was just the first four or five months. At eight months we found out he had an intestinal malrotation and needed surgery for that as well as a nissen fundoplication and g-tube placement. That was the first of many intestinal surgeries and hospitalizations. At age three there was another suspicion that was confirmed —he also had autism. We spent so much time at Maine Medical Center in Portland, Maine, that he and I virtually lived there. My husband and our older daughters, ages 11 and 14, shuttled back and forth to “visit” us. There were so many doctor and therapy appointments, so many evaluations and tests, procedures and surgeries that about four or five years passed in a blur. See, I don’t even know how many years!
But gradually Sam was better intermittently, and we adjusted to our new life. He eventually sat up, crawled, and learned to walk with a walker. He did not learn to talk, and he still had a lot of medical problems and developmental delays. He had insomnia. It turned out that he wasn’t blind in the ordinary sense, but he did have cortical visual impairment. It was pretty clear that he was never going to be very much like any other kid. He had very strange, maladaptive behaviors and a lot of screaming when things weren’t the way he thought they should be (like, if the car turned right and he liked left turns). It was pretty clear he was not going to be very much like any other kid. There was a lot to be scared about.
He started “home” school at a few months of age — vision therapy, physical therapy, occupational therapy, developmental therapy, and then some kind of autism therapy. By age 2.5 he was off to special preschool in the mornings and the individual therapists came in the afternoons. Too much therapy!! But at age 4 we began a new kind of therapy, Applied Behavior Analysis — at school and at home. That was a great turning point. We finally had everyone on the same page, working as a team. And if they didn’t want to get on board, they got out. We were learning too.
About the time Sam was turning five, our family made some decisions. It was pretty clear that I couldn’t work outside the home because there was no daycare or provider for Sam. His doting sisters were now 19 and 16 and it was pretty clear that Sam was the center of the universe to all of us…was that the best thing for him? For us? We had the bright idea that we could put our newfound skills to work, give me a “job” so I wasn’t so totally focused on Sam, and provide siblings to Sam nearer his age. A win-win!! The job? We became a treatment foster family. This means that we provided a home to kids with developmental or mental health issues.
Ho ho ho! and it wasn’t even Christmas! First came a couple of sisters aged 11 and 12 — not exactly the little boys Sam’s age that we had pictured, but they definitely became siblings, made our lives more lively, and kept us from over-focusing on Sam. They stayed with us until they were ready to spread their wings and fly. Next came another girl…this time only six years old, then a few years later a girl aged 11. Again, these kids were permanent siblings — Sam, and the two younger girls were each only a year apart in age. We had many others in and out temporarily while they were either working on returning to their birth families or moving on to a different situation, or just for regular respite.
So Sam was never lacking for siblings…especially sisters. It wasn’t always easy, it wasn’t always pretty, but it was our family. It was a hopping household for sure.
Meanwhile, Sam grew, and grew older. He had his intermittent emergency hospitalizations, usually related to his gastro-intestinal abnormalities. He had a few surgeries, lots of doctor appointments, lots of testing, some ups, some downs. When he was 10, we had to leave our hometown due to the paper industry going belly-up…from human resources in a very large paper company, David moved to human resources in a small hospital, and we moved 50 miles down the road. We decided it would be a good move and adjusted to new providers, a new school system and a new house. And good news! We were now 45 minutes closer to the nearest service town! Since we have always had to make that trip to Bangor at least once a week and sometimes daily, that was a big deal. And the ambulance could get to Portland 45 minutes quicker too…always look on the bright side of life!
And so time passed. Sam went to school — there were ups and downs. He developed an exaggerated startle reflex, hyperekplexia, which further compromised his ability to walk. We spent untold hours advocating at the state and local level for appropriate services. We had a few unpleasant experiences and we had a lot of pleasant experiences. He had some wonderful teachers and after-school staff, he had a few not-so-good ones. We traveled a lot: by planes, trains, and automobile and had a lot of amazing adventures with our crazy family. Sam never learned to walk independently or to talk, but he learned to get around and he figured out how to communicate in his own way. (He had to with all those sisters!) He made friends, oh boy, did he make friends! Time flew by, and then it was time to cross over into the dreaded “other side”…adulthood.
And you know what?? It is great here!
Bored? Lonely? Stuck at home? Never!! Over the past few years Sam has truly found his place in life.
One advantage of living in a very rural area is that there are no “day programs” —Sam has one-on-one staff for 37.5 hours a week. They are busy — with Special Olympics, their self-advocacy group, friends, and community groups all over town. He has one special volunteer job, and participates in lots of other special projects, for instance, they are planning a special KAT6A fundraiser for spring, because Sam finally has a diagnosis, and a cause, and his friends want to help. Sam has more friends than I do!
He still lives at home with us because that is what we choose. And because we find it more difficult to travel now that Sam is a grown man, we have turned our property into a full-fledged redneck resort, just for Sam and our family and friends. If we can’t go to them, we want them to want to come to us, and they do come! Three of his sisters left home, went out to explore the world, and then came home to Maine. Along the way they found and married three very special men, who love Sam almost as much as his sisters do. Another sister lives in a nearby town. There are children, dogs, babies, friends, and family underfoot constantly…and adventures to be had with all of them. Uncle Sam is a great favorite — who else’s uncle can play with toys the way he can?? And Sam has lots of fun grown-up toys everyone wants to share!
He makes a difference to many and has a fulfilling, meaningful place in his family and community. His greatest gift is making people feel loved—his smile can light up a room and his hugs make troubles melt. He participates. He contributes. There isn’t anyone who knows Sam who doesn’t love Sam. What more could anyone want?
It is not the life I pictured when I married my handsome prince nearly 43 years ago. But it is a far cry from the life I feared and worried about twenty years ago.
Life is good here on the other side with Sam.
Filed under: Blog
Comments: Comments Off on Life on the Other Side
When I first discovered that I was pregnant with Savannah at just 8 weeks something immediately felt off. Having a child before I found myself questioning that I didn’t feel any clear signs or symptoms of pregnancy. I remember thinking that something might be wrong and that unfortunately my body may reject this unexpected and precious gift. I worried and wondered until we had our first OB appointment at 13 weeks. When I saw her little heartbeat I was so relieved!
Just when we thought we were in the clear the ultrasound tech said she had to bring in a doctor. My heart sunk and I knew in that moment that my instincts had been right. There was a problem with our growing baby. The doctor came in and kept quiet as he circled the same area of my abdomen over and over again. He finally looked at my husband and I and said, “one kidney is extremely large, much larger than it should be at this time.” He explained nothing further and said we would need to be followed by a maternal and fetal health specialist.
This is when our journey really began. We followed up with the suggested specialist where we were informed that she had “hydronephrosis” essentially a birth defect that was causing her to retain urine in her kidney. We were informed that she had a 30% chance of having other birth defects as well that may not be seen via ultrasound. We were asked if we wanted an amnio and then the dreaded topic of abortion was presented. We denied both. There was lots of talk of a premature delivery or the possibility of inducing me early to perform emergency surgery on her kidney. A ton of worry, stress and fear had fully taken over. We continued to see our specialist along with our regular Ob-Gyn over the remaining course of the pregnancy. I had more ultrasounds and non stress tests than I could keep track of.
I went into premature labor at 28 weeks and again at 32 weeks, I was given medication to reduce contractions and shots to mature Savannah’s lungs; just in case we couldn’t keep her in any longer. Luckily she stayed put just long enough to make it full term, 37 weeks ! She entered the world at 6 lbs. 3 oz. She was BEAUTIFUL!
During her initial physical it was noticed that she had a cleft palate, microcephaly, bilateral Simian creases and several other markings that appeared to be syndrome like. But strangely for the first time since finding out we were expecting, I had no fear. I looked at her beautiful little face and I felt at peace. I knew that we had a lot ahead of us but I also knew that we would be ok. She had some breathing difficulty and was refusing to eat, so she was transported from her birth hospital to a major medical facility several hours away.
We met a team or physicians in the nicu, a geneticist, a cardiologist, a pulmonologist an ophthalmologist, and a neurologist. They immediately began to test Savannah for every possible syndrome they thought she might have. Test after test came back negative or normal. We had an extremely hard time with feeding, and she just wouldn’t eat. A therapist came in to work with her do to her cleft palate, but still no luck. It was at that point that a gastroenterologist wanted to perform a scan to make sure that her everything was okay with her intestines. Her results came back that she had a malrotated intestine. (another birth defect)
At 2 weeks old, Savannah went through surgery to correct this problem and have a G-tube placed. We spent another 6 weeks in the NICU before being discharged with no answers. After Savannah came home she started to have serious blue spells, she was re-admitted to the NICU for an additional 2 weeks to run further testing where it was discovered that she had severe reflux. With anti-acid medication and a pulse ox monitor we were finally going home.
Weeks and months were flying by and all although Savannah wasn’t growing much she seemed healthy. We continued to have follow-up appointments with all of her specialists and tested for new syndromes with every chance we got. Months turned into years. Savannah had severe developmental delay and did not sit unsupported until she was 18 months old. She began to crawl at 2 and 1/2 years old and pulled to stand for the first time at age three. She was seeing therapists multiple times a week for feeding, speech, occupational and physical therapy.
When Savannah was three and a half years old she woke up late one morning (not like her), she appeared to be getting sick, no fever but she looked sleepy and was occasionally gagging. In just a few short hours it was clear that Savannah was in serious distress and something was terribly wrong. We rushed her to the ER, she had to be transported to a nearby hospital via helicopter where it was confirmed that she had a large bowel obstruction. Her body was going through septic shock and she had to be put in a medically-induced coma immediately. She was sent in for emergency surgery where they corrected the obstruction and resected several inches of dead bowel. She required 3 blood transfusions and was left open for 24hrs before they had to take her back into the OR and & resect yet again. She had an extremely hard and difficult recovery ahead of her. She was kept in a sedated coma for 3 weeks total and against all odds was able to fight through the worst time in her life and come back stronger than ever!
A few months later at our annual follow up with our geneticist she had mentioned that she would like to try to get Savannah approved for whole exome sequencing. It was through this test that a diagnosis was finally made! 4 years later! Savannah’s test had come back with a spontaneous nonsense variant in her kat6a gene. At the time we were told that she was the second person EVER to be diagnosed and we had no idea what it meant or what to expect. We agreed to have her results and findings published in hopes that others undiagnosed wouldn’t have to go through years of not knowing.
We researched Kat6a endlessly, we tried to find anything and everything we could online and in medical books, but got nothing. 2 years later we finally found a website randomly on the internet about a child with kat6a! Finally we weren’t alone! We reached out to the family and to our amazement we discovered that there was a Facebook page where families who had loved ones diagnosed were coming together! Several more diagnoses had been made! It was astonishing to see the similarities in the facial features of our children and to hear how much they were all alike. Little by little our group was growing.
Our published papers and reports were working, our consent to make Savannah’s results from exome sequencing public had allowed other families to find hope and answers as well. Today we are still searching for help, understanding and treatment. Not much is known about kat6a but we are now at one hundred and seven people strong and we will not give up!
Savannah is now seven years old, she still has many difficulties in life but with perseverance comes progress. I’m thrilled to share that she just learned how to walk independently this year! She is a complete Joy to be around, so easy going and happy all the time. We couldn’t imagine our lives without her and without doubt we are better people because of her! Looking back on Savannah’s story isn’t easy she’s been through more tests, procedures, surgeries and obstacles than anybody I know. As her parents we have had to face plenty of heartache, frustration and anger along the way but who doesn’t!? Life is a beautiful struggle sometimes and as long as we can help just one person to find hope, and happiness through her story then it’s worth reliving it a million times! We try to take things one day at a time now and try to live each day to the fullest ; instead of living in fear of the unknown. We are so proud of savannah, how far she’s come and all of her accomplishments and we look forward to all the happiness and triumphs for her that are still yet to come.
By Lindsey
Filed under: Blog
Comments: Comments Off on Sweet Savannah, Second Diagnosed
Franki was born on Mother’s Day 2016. Shortly after he was born the paediatrician informed us that he had been born with a few birth defects that, by themselves, shouldn’t amount to anything to warrant us to worry about. But we did. We were shattered and overcome with anxiety. Little did we know then, that it would take an 18 month journey of worry, uncertainty and frustration before we would receive the life-changing diagnosis of KAT6A Syndrome.
Franki’s fight began 5 days after birth when we were told by his craniofacial surgeon that Franki would need to have major cranial surgery to repair his sagittal Craniosynostosis at 3.5 months of age. Before we could schedule this in, Franki underwent emergency surgery to repair an inguinal hernia at 4 weeks of age and in doing so they also performed an Orchiopexy to correct his other Cryptorchidic birth defect.
By 6 months of age, he had missed several important milestones such as being visually attentive and tracking objects or recognising faces, tolerating tummy time, rolling over and sitting up. Franki’s hands were also permanently fisted, held up to his face and he regularly arched his back and twisted his legs – all red flags for what we first feared may have been due to Cerebral Palsy, but later discovered was due to something called Dystonia, a neurological muscle movement disorder.
Looking back, we knew from the very start that there was something very wrong with our blue eyed baby boy. Breastfeeding Franki proved immensely difficult, made worse by a double tongue and lip tie. He had severe reflux (and still does) which we sought medication for, suffered terrible colic and constipation (now managed by daily medication) and cried incessantly all day every day for much of his infancy, so much so that we nick named him Cranky Franki during these episodes.
We received the heart breaking news that Franki had cerebral vision impairment after first suspicions of delayed vison maturation had not improved by 7 months. A very poor sleeper by nature, Franki’s irritability grew, much to the mystery of his doctors at the Princess Margaret Hospital for Children here in Perth.
By 8 months of age, and after many trips to the hospital and various doctor’s appointments later, Franki was diagnosed with global developmental delay and so started his multi-disciplinary therapy journey and our first appointment with the genetics team followed soon after.
AT 15 months of age, Franki had his first 7 minute seizure. He underwent many tests including CT’s, MRI’s, EEG’s, a lumbar puncture, ultrasounds and x-rays. All genetic tests also came back unremarkable and within normal limits. Fast forward to November 2017, the Genetic Services of Western Australia had almost exhausted all testing available to them until they detected a pathogenic variant on his KAT6A gene from a Massively Parallel Sequencing via TruSight One test. We then learnt that Franki was only the 107th person in the world to be diagnosed!
Aside from his many challenges, Franki is a very affectionate and warm natured little boy whose smile, is pure magic, and whose determination to succeed is inspiring to all that know him. Currently, Franki is still non-verbal, but is now sitting independently at 18 months and continues to surprise us every day. Whilst his diagnosis is bittersweet, it is validation for the concerns we suspected from the very beginning, and ultimately, it has enabled us to forge a strong connection with other KAT6A families who share the same unique journey, all over the world! Pretty special, indeed.
By Vera
If you are interested in learning more about Franki’s story, visit http://frankijulesmoura.net/
Filed under: Blog
Comments: Comments Off on Franki the Fighter
Will was born in 2015 at 41 weeks and weighed a healthy 7 pounds, 12 ounces. We thought he was the strongest baby in the world because he held his head high immediately after delivery. I have the most amazing photos of him, minutes old, laying on my chest pushing his own chest and head up. It was impressive to my husband and the delivery nurses, but I know now that should have been our first red flag. In the week following, whenever he was placed on his tummy, he would roll over. We were so proud of our super baby and we joked he’d be walking by nine months. When I mentioned this to his pediatrician, she seemed to think it was a fluke and that this rolling wouldn’t persist. Breastfeeding was a huge struggle. It was extremely difficult to get him to latch since his body refused to relax, but I chalked it up to being normal since not all babies are natural breastfeeders and I succumbed to exclusively pumping when his pediatrician wasn’t satisfied with his weight gain. By three months it was evident that his muscle tone was much higher than it should be. His hands were permanently fisted and creases had formed by his elbows and knees since he kept his body scrunched up at all times. It was nearly impossible to stretch out his body for an accurate length measurement. That’s when we first discovered the term hypertonia, which would begin our journey to finding answers.
At four months we were referred to a neurologist. The first neurologist did an EEG and it was normal. He found his head percentile to be less than the tenth percentile, but that was not concerning. Since his other milestones were on pace, such as smiling, laughing and eye contact, we held high hopes that through some physical therapy that he would loosen up. After an evaluation by our local Early Intervention program, he was disappointingly denied services since his high muscle tone had not resulted in a delay in any of his milestones. In fact, he was able to sit for a few seconds on his own. So I was left with some daily stretches to do with him.
At five months my concerns grew and I decided it was time to get him physical therapy privately, so I called the physical therapist who had done his initial evaluation. At this point he had stopped rolling over and wasn’t tracking objects and was having intense sleep issues since his body just couldn’t seem to settle. Upon my first session of private therapy, the physical therapist noticed his regression immediately. She expressed great concern for my little boy and recommended that I see a different neurologist and she personally requested a new evaluation through Early Intervention since she had no doubt he would now qualify.
By six months, Will’s head percentile had fallen to the third percentile. The new neurologist that we saw prescribed a brain MRI and an array of blood work due to his poor reflexes, hypertonia, poor visual tracking, gastrointestinal issues, difficulty feeding and microcephaly. The results of the MRI and all the blood work were normal. So the next step was to see a geneticist to rule out any rare genetic disorders. The initial genetic testing all came back normal including the microarray test. We were thrilled and very hopeful that Will would outgrow these symptoms since each specialist’s general impression of him was fairly normal and we were repeatedly told that “he looked good.”
At a follow up visit with the geneticist at nine months, he suggested that we do Whole Exome Sequencing since our insurance agreed to cover its cost. In the several months that we waited for test results, Will’s development began to improve. His head size stayed at the same percentile, he was tracking objects, sitting independently, playing with a variety of toys in his jumper, swallowing purees and began crawling just after his first birthday. So at fourteen months when my husband and I were given the KAT6A diagnosis, we were extremely shocked and devastated since we had convinced ourselves that the test results would come back normal as they had in every other instance. At that time we were told that there were only a dozen others in the world with this diagnosis and that they were nonverbal, profoundly intellectually disabled, and in most cases had heart conditions. We were told to see cardiology immediately and to think about signing in the future since the literature stated that some of the children communicated that way. It felt like our world had come crashing down. It had never dawned on us that our baby may never talk, learn to read, live independently or likely have serious health problems.
In the following days and weeks, I read anything I could get my hands on and was determined to understand the science behind it. After cardiology ruled out any abnormalities, I felt some anxiety lift. I dedicated my time to making sure that he would receive the highest level of early intervention therapy that was available, and so I requested speech/feeding services and small group therapy in addition to the PT, OT and teacher he was already receiving. My husband and I also did a lot of grieving and worrying during this time. You name it, we worried about it.
Two months after D-day, as I sometimes refer to it, I decided to reach out to the email address on the Chloekat6a.org website. That’s when I learned about the Facebook support group and my life forever changed. I found on the support group that Will was actually the 40th child diagnosed and that there was even a mother of an adult in our group. I cried as I watched videos of children riding their bikes, swimming in pools, fluently signing, talking and even doing math. This page portrayed so much love and hope that I could never have gotten out of reading technical research articles. Despite a huge range in abilities, I could see evidence of joyful kids every where and that is when I knew that this diagnosis would not define Will or our family’s happiness. I was truly overwhelmed by the number of people who reached out to me to share their stories, offer information and emotional support.
Through the support group, I was connected to Dr. Richard Kelley, and under his advice started Will on a mitochondrial cocktail shortly after his second birthday. The effects of the cocktail were noticed almost immediately in Will. His stamina and energy increased, constipation improved, he began eating solid foods rather than solely purees, and within weeks began walking independently.
Today, Will is a healthy 2 1/2 year old. He is the sweetest, happiest, most lovable boy with global developmental delays. He is very active and into everything. Many of Will’s developmental issues relate to motor planning difficulties and some lingering muscle tone issues. He is a solid walker and climber and is working on walking up/down stairs independently. He has four signs that he uses regularly to communicate and a few words that he uses from time to time. His receptive language is significantly better than his expressive. He loves to identify body parts and follows simple commands, such as “put your bottle in the sink.” He has some difficulty eating, but this has improved greatly with the help of a feeding therapist. Currently he eats a variety of foods cut into small pieces, and we are working on tearing pieces of food with his teeth himself, such as taking a bite from a slice of pizza. He has fine motor delays and does not have a proper pincer grasp. He has some sensory issues, but they are manageable. He mouths objects frequently, hates having his teeth brushed, covers his ears at loud drawn out sounds such as the vacuum, loves movement and is very stimulated by light. He sleeps well at night, but naps inconsistently. He is beginning to develop a playful relationship with his five year old brother, which is beautiful to see.
Parenting Will has allowed me the greatest highs and lows of my life. He has taught me to rejoice in the little things that I took for granted with my older son. He has amazed me with his persistence and determination. And although he cannot say it, he shows me so much affection in his tender touch and nonverbal request for a kiss. His laugh is contagious and everyone who gets to know him can’t help but fall in love his sweet soul.
By Aimee
Filed under: Blog
Comments: Comments Off on Live, Love and Laugh with Will
Coucou..je m apelle Chloé..j ai 16ans et demi
Maman va vous raconter un bout de mon histoire
Il a fallut 16ans et 5 mois pour enfin trouver le nom de la maladie: KAT6A je suis là seule à ce jour diagnostiquer en Belgique
16 longues annees de combat… a frapper à toutes les portes.. un parcours du combattant…de douleurs..de peur..de doute..de pleurs…d angoisse et d impuissance et surtout seules face à ses médecins qui ne comprenais rien..
Un merci tout particulier à mon généticien qui n a jamais baisser les bras et à la cardiologue qui nous a jamais abandonnée..et a mamy et papy
Mais nous n avons jamais perdu espoir
Chloé est une vraie petite guerrière..a traversée déjà beaucoup trop d opération et de souffrances du haut de ses 16 ans
Son sourire..sa force..sa joie..son courage..son amour..sa persévérance.
Une vraie merveille
Mon diamant! mon trèfle à 4 feuilles,unique elle est extra-ordinaire.
Née à 39 SA..2kg820..TN:47cm..
PCN:31,5cm
-Microcephalie pré et post natale
-apnee du sommeil
-Assise à 2 ans..et la marche à 2ans et 8mois
Épilepsie
-Absence de language..a ce jour dis quelques mots une trentaine et le plus beau “maman Je t’aime” a 15 ans et essaye de se faire comprendre
-Artere sous claviere droite rétro oesophagienne
-RGO important
-difficulté alimentaire avec nécessité de gastrostomie et nyssen a l âge de 2 ans
A ce jour Ne mange plus du tout à part du liquide par biberon ou à la paille
-intolérance aux bruits
-déficience intellectuelle retard mental
-bruxisme important
-sommeil agité et très difficile
-Opérer d une CIA a 5ans
-fuite valve mitrale
-Hypotonie
-amyotrophie
-atrophie musculaire ne pèse plus que 38 kilos
-Pieds et mains toujours froids
-Hyperlaxite articulaire douleurs intenses dans les membres:dérivé de morphine
-coudes inversé
-Cyphose dorsale
-scoliose
-instabilité des rotules luxables
-troubles du comportement
-Dysfonctionnement du tronc cérébral responsable de trouble de la fonction alimentaire majeurs
-Opérer 2 fois des dents trop petite mâchoire
-anévrisme de l aorte ascendante stade 3
Prise de betabloquant
-constipation sévère impossible à gérer
-ventre toujours gonflé et remplis d air
-dépendante de moi pour les gestes du quotidien:habillage,toilette,manger
– a besoin d une chaise roulante pour ses déplacements car plus de tonus musculaire..fatigue et douleurs
Chloé a aussi une profusion de naevus plus de 100
Des exéreses ont déjà été réalisé sur plusieurs car ils étaient très suspects heureusement ils étaient benins
A ce jour elle dois encore se faire opérer de plusieurs naevus qui semblent très dangereux
Mon généticien pense que j ai aussi une autre maladie qui engendrerait une profusion aussi importante
Voilà vous savez les grandes lignes
Je voulais juste remercier ce groupe..ces parents incroyable et ses petits héros qui sont devenu comme une famille..un repère..un phare qui m’éclaire.
Par Carine
English Translation:
Hello..I call myself Chloe..I am 16 years and a half Mom will tell you a piece of my story It took 16 years and 5 months to finally find the name of the disease: KAT6A I am here alone to date diagnose in Belgium 16 long years of fighting … knocking on all the doors .. an obstacle course … of pain … of fear..of doubt … of tears … of anguish and impotence and especially alone in front of his doctors who did not understand anything .. A special thank you to my geneticist who never give up and to the cardiologist who has never abandoned us … and has mamy and grandpa But we have never lost hope Chloe is a real little warrior … has already gone through far too much surgery and suffering from her 16 years His smile, his strength, his joy, his courage, his love, his perseverance.
A real wonder My diamond! my clover with 4 leaves, unique it is extra-ordinary.
Born at 39 SA..2kg820..TN: 47cm .. NCP: 31,5cm
-Microcephaly pre and post natal
-Sleep Apnea
-Seated at 2 years old..and walking at 2 years and 8months Epilepsy
-No language..a date say a few words about thirty and the most beautiful “mom I love you” at 15 years old and tries to be understood
-Arte under right retro esophageal keyboard
-RGO important
-difficult food with need for gastrostomy and nyssen at the age of 2 years. To this day does not eat anything other than liquid by bottle or straw
-noise intolerance, mental retardation
-bruising easily
-sleep agitated and very difficult
-Operate a CIA at 5 years
-passed mitral valve
-Hypotonia
-amyotrophie muscular atrophy weighs only 38 kilograms
-Feet and hands always cold
-Hyperlaxitis articular intense pain in the limbs: derived from morphine Inverted
-Cyphosis dorsal
-scoliosis
-instability of luxable patella
-troubles behavior
-Brainstem dysfunction responsible for major food function disorder
-Operate teeth twice too small jaw aneurysm of ascending aorta stage 3 Betablocking
-serious constipation unmanageable
-always inflated and filled with air
-dependent on me for the daily gestures: dressing, toilet, eating
– needs a wheelchair for his movements because more muscle tone … fatigue and pain
Chloe also has a profusion of more than 100 nevi Exereses have already been made on several because they were very suspicious fortunately they were benign. To this day she still has to undergo surgery of several nevi that seem very dangerous. My geneticist thinks that I also have another disease that would generate such a large profusion. Here you know the main lines. I just wanted to thank this group..this incredible parents and her little heroes who have become like a family..a landmark..a lighthouse that enlighten me.
By Carine
Filed under: Blog
Comments: Comments Off on Chloe’s History
Our sweet son Holden was born in April of 2015. He was diagnosed with KAT6A in December of 2016. The doctors told us there were issues present during the pregnancy. Holden had enlarged kidneys and calcium deposits on his heart and bowel. We saw a few specialists and had some tests ran but nothing showed any major problems. I was also diagnosed with oligohydramnios (low amniotic fluid) due to his kidneys during the second and third trimester of my pregnancy with Holden. He was delivered emergency C-section at 36 weeks at our local hospital. We noticed right away that he was having a hard time sucking therefore he couldn’t eat, he also was very small only 4lbs 14oz. The doctors decided it was best that he be treated at a neonatal intensive care unit a few hours away. That was the hardest thing as a mother that I have ever had to do, watch helplessly as my precious newborn baby was rushed in an ambulance to another hospital so far away. He spent about 5 weeks in the nicu. They ran so many genetic tests on him and everything came back normal. While he was in the nicu we learned that he had hydronephrosis of the right kidney, his testicles were ascended, and he had mal rotation of his intestines, among other small issues such as hypertonia, hypotonia, low set nipples and ears, bilateral ear pits and a bicuspid aortic valve. Holden also had to have a g-tube insertion and surgery on his intestines due to the mal rotation.
He got to come home from the nicu when he was around 1 month old. We were set up with many specialists to see over the next year including cardiology, neurology, plastic surgery, neuro surgery, G.I, urology and genetics, hoping to find out what was causing Holden so many issues. When Holden was a couple months old we learned that he has a rare eye condition called CVI, he also has astigmatisms and is near sighted. Holden was also diagnosed with microcephaly and a neck condition concerning his c1 and c2 vertebrae. We got set up with our local CDSA agency and now have awesome therapists to work with. Holden receives P.T., O.T., vision and speech. When we saw our genetics team when Holden was just a few months old they set us up with a research program that offered whole exome sequencing. It took about 6 months but they gave us the answer we had been looking for, for almost 2 years, that Holden had a rare syndrome called KAT6A.
Holden has had 3 surgeries since birth, the mal rotation of his intestines, deflux of his right kidney, and had his testicles descended surgically. He will have one more surgery this year on his neck. He is going to have his c1 and c2 vertebrae fused to his skull because they did not form properly. We pray that will be his last.
Although Holden has been faced with so many challenges medically and physically he has accomplished so much. He was sitting at 16 months and beginning to eat pureed foods. Now that he is going on 3 years old he is crawling all over the place and chasing his brothers, and he is eating all kinds of pureed foods with texture added. We are so very proud of him! It is truly amazing to us what this sweet boy is capable of and we know as his family that the sky is the limit. He is the sweetest most loving child I have ever met. He loves his two older brothers and our dog. He loves bubble baths and funny noises. Holden just lights a room with his smile. We are very confident that he will walk and communicate with us through signs and/or verbally.
By Brittany
Filed under: Blog
Comments: Comments Off on Sweet Holden
Madison is 3 years old and was diagnosed with KAT6A a few months ago.
She is non verbal and doesn’t walk yet. She does shuffle her bum along the floor very fast and holds on to things to get around. She is the happiest little girl on the planet. She has low muscle tone and a hole in her heart. She also has severe gastro, which she takes omeprezol for everyday and is on melatonin for sleep at night. Her older brother and sister love spending time with her as do we. She started nursery school in September and is loving it. She is a joy to the world.
By Tony
Filed under: Blog
Comments: Comments Off on Joyful Madison